
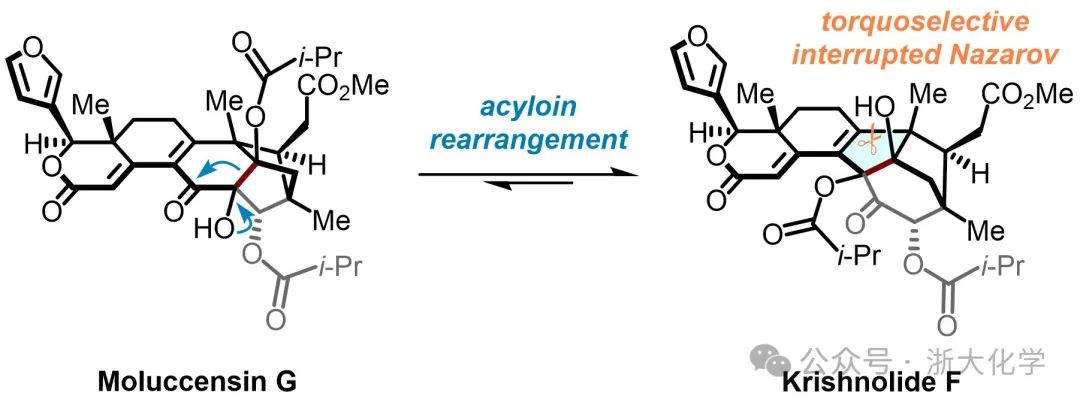
近日,我系丁寒锋教授与宣军团队合作报道了两个phragmalin型(moluccensins G和H)和两个khayanolide型(krishnolide F和khayseneganin F)柠檬苦素类天然产物的发散型全合成,发表在JACS上。在这项研究中,作者以扭矩选择性的中断型Nazarov环化反应为关键步骤,结合Liebeskind–Srogl偶联、安息香缩合和双向偶姻重排等核心转化,完成了分属两种不同骨架的四个柠檬苦素类天然产物的首次不对称全合成,同时也为phragmalin型与khayanolide型柠檬苦素之间的生源转化关系提出了新的见解。
柠檬苦素类天然产物(以下简称为柠檬苦素)最初源于存在于柠檬或者其它柑橘属植物中的苦味成分,它们广泛分布于楝科、芸香科和苦木科植物中。其骨架结构和氧化模式复杂多变,可据其结构进一步被划分为三种类型:环完整型柠檬苦素、环断裂型柠檬苦素以及环重排型柠檬苦素(Figure 1),后两类是由4,4,8-三甲基-17-呋喃甾族前体经一系列氧化裂环和骨架重排所得。除了具有错综复杂的结构外,柠檬苦素还表现出了包括抗癌、抗炎、抗疟和抗菌活性等在内的良好生物活性。自Corey课题组于1989年首次完成azadiradione的全合成起,合成化学家们对于柠檬苦素的研究兴趣逐步增长。近二十年来,已有国内外十余个课题组在此领域做出了突出贡献。
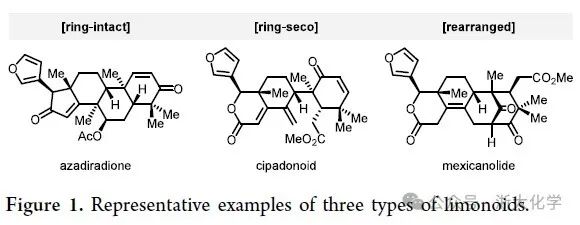
Moluccensins G和H(1和2, 见Scheme 1)是从印度红树Xylocarpus moluccensis的种子中分离得到的两个phragmalin型柠檬苦素,其结构上除了具有该家族特征的三环[3.3.12,10.11,4]奎烷骨架外,还含有一对△8,9和△14,15共轭双键。近期分离得到的krishnolide F(3)和khayseneganin F(4)具有特征的三环[4.2.110,30.11,4]奎烷骨架,属于khayanolide型柠檬苦素,与phragmalin型柠檬苦素在生源合成上具有一定的相关性。初步研究表明,krishnolide F(3)具有促孕甾烷X受体(hPXR)激动的生物活性。值得注意的是,khayseneganin F(4)紧凑的笼状六环骨架中嵌有十二个连续的手性中心,其中六个处于桥头位置(三个为全碳季碳),其合成具有高度的挑战性。
Nakatani等人曾提出phragmalin型和khayanolide型柠檬苦素生源关系假说:phragmalin型柠檬苦素可经自由基重排或频那醇重排途径释放环张力,驱动C1(2→30)重排,完成向khayanolide型柠檬苦素的转化(Scheme 1B)。然而,该假说的合理性尚未得到充分阐明,且自然界中C1在C2和C30位点之间的迁移倾向也仍不明确。尽管在柠檬苦素的合成研究领域已取得了诸多进展,但合成化学家们对具有独特骨架的phragmalin和khayanolide型柠檬苦素的合成探索仍涉足甚少。目前仅有Sarpong和Jirgensons课题组报道过相关的合成研究,均主要聚焦于phragmalins的三环[3.3.12,10.11,4]奎烷笼状骨架(A1A2B)的构建,未能完成相应天然产物的全合成。
为了进一步探究phragmalin型和khayanolide型柠檬苦素的生源关系,作者选择了moluccensins G和H(1和2)、krishnolide F(3)以及khayseneganin F(4)作为合成目标,并提出了如下逆合成分析(Scheme 1A):作者认为逆生源合成假说的C1(2→30)的重排反应在化学上亦是可行的,于是将五环中间体5选为共同中间体,其经偶姻重排或官能团转化即可实现phragmalin型柠檬苦素1和2或khayanolide型柠檬苦素3和4的发散性全合成。而5中的A2环可由四环醛酮6经分子内的安息香缩合反应构建。中间体6的中心B环可由二烯酮7经中断型Nazarov环化反应构建。于此,作者希望通过底物自身控制,完成B环构建的同时实现C1位叔醇和C10位季碳两个手性中心的立体选择性引入。中间体7可由硫酯8和烯基锡化合物9经Liebeskind−Srogl偶联拼接得到。
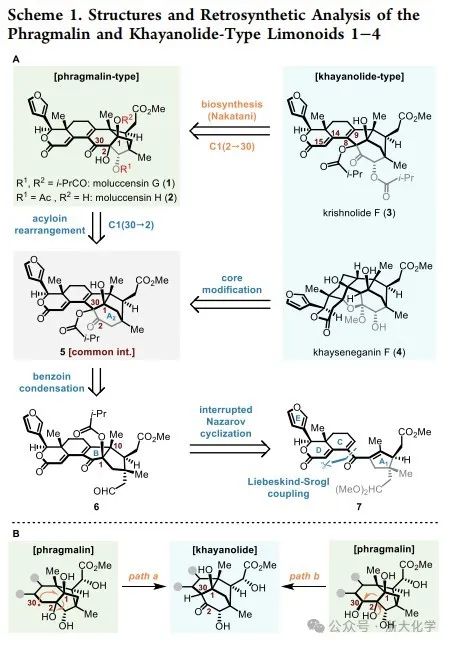
首先是硫酯8的制备(Scheme 2A)。作者以商业可得的手性烯酮10为起始原料,使用乙二醇和p-TsOH·H2O使其发生缩酮化和双键迁移,再利用Upjohn双羟基化反应制得邻二醇11。邻二醇11与原位制备的Ph3PCl2在82 ℃下反应,经半频哪醇重排过程可生成具有6/6反式并环结构的酮12,其经一锅法的生成三氟甲磺酸烯醇酯和CuI催化的交叉偶联反应引入甲基可转化为烯烃13。烯烃13经臭氧化断裂双键可得不稳定的醛酮中间体(未展示),其立即被投入AcOH/哌啶介导的羟醛缩合反应,最终获得缩环产物醛14。随后,醛14经氮杂环卡宾(NHC)催化的氧化硫酯化反应和区域选择性的烯醇醚化反应生成16。烯醚16经选择性臭氧化断裂Δ2,7-双键以及缩醛化反应即可以得到硫代酯8。
随后是烯基锡化合物9的制备(Scheme 2B)。6-甲基环己-2-烯-1-酮(17)先与3-呋喃甲醛发生非对映选择性aldol反应,加成产物经α-碘代反应可得到中间体18。接着,作者经过大量的条件筛选,发现采用手性碱(R)-CF3-BTM(19)和(BrCH2CO)2O的组合可以理想的效率和对映选择性实现了二级醇18的酰化动力学拆分,得到溴代乙酸酯(+)-20。在后续内酯D环的构建过程中,溴代乙酸酯20在常规的Reformatsky反应或锂卤交换条件下均伴随大量的脱卤副产物,导致环化收率不佳。最终作者采用PhTeLi(由碲粉和PhLi原位制备)和CeCl3的条件成功实现了预期的关环。所得产物(未展示)与Me6Sn2偶联即可完成烯基锡9的制备。随后,片段8与9在催化量Pd2(dba)3和AsPh3以及化学计量的二苯基膦酸铜(CuDPP)的作用下发生Liebeskind–Srogl偶联反应,以97%的产率顺利地得到关键反应前体21,其三级醇经消除即可获得另一可能的关键反应前体7。
接下来是对中断型Nazarov环化反应的研究(Scheme 3)。作者首先尝试了一系列经典的酸促Nazarov环化反应条件,然而均未能实现期望的转化。因此作者转而尝试更温和的光照促进的Nazarov环化反应。令人沮丧的是,二烯酮7在i-PrCO2H和254 nm紫外光激发下,经光促烯醇互变转化为Δ9,11,Δ8.30-二烯醇22后,随即发生6π电环化得到四环副产物23。为了抑制该副反应的发生,作者采用C14位羟基未消除的二烯酮21在相同的条件下进行反应。此时Nazarov环化反应顺利发生,得到了一对比例为4.8:1的混合物24与25,总产率为75%,其中所需的非对映体25仅占少数。为认识并改善这一不理想的扭矩选择性,作者对Nazarov环化的反应机理进行理论计算的研究(Scheme 3B)。计算表明,在不期望的环化路径中C14-OH与羰基之间存在着明显分子内氢键缔合,该氢键作用可能一定程度上稳定化了这一路径(ΔGTS1_OH−TS2_OH = −0.7 kcal/mol)从而导致不期望的立体选择性。由此可推测,如果使用合适的基团掩蔽C14-OH破坏分子内氢键,这一不理想的扭矩选择性可能有望得到逆转。进一步的底物筛选显示,由21经甲基化制备的甲基醚21′在光促Nazarov环化条件下能以76%的收率单一的立体选择性转化为预期的环化产物25′。该结果亦可从模型过渡态TS1_OMe中C14-OMe与酮羰基间显著的偶极排斥(ΔGTS1_OMe−TS2_OMe = 6.1 kcal/mol)得到合理解释。
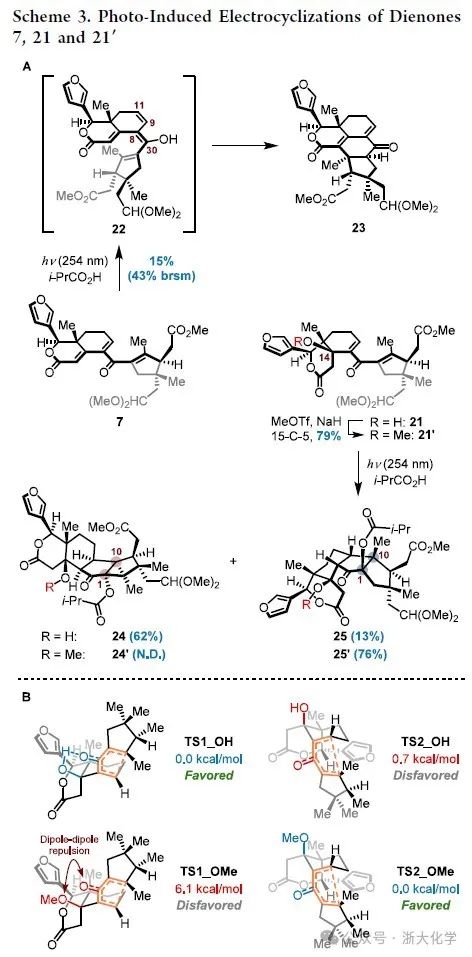
顺利获得Nazarov环化产物后,作者开始着手共同中间体5的合成(Scheme 4)。高级中间体25′在K2CO3和DDQ的条件下发生碱促消除反应和氧化脱氢反应构建共轭二烯片段,随后在p-TsOH·H2O的作用下释放醛基,最终以65%的收率得到安息香缩合前体6。作者经大量的条件筛选发现,6的安息香缩合反应在反应体系严格脱气的情况下,使用三唑盐26作为NHC前体与NaOAc作为碱的组合可取得最优的结果,最终以51%的收率获得共同中间体5。值得注意的是,此时伴随的异丁酰基从C1位叔醇至C30位叔醇的迁移是自发的,控制实验表明该过程的耦合极大地推动了安息香缩合产生的Breslow中间体(未展示)向环化产物5的不可逆转化。
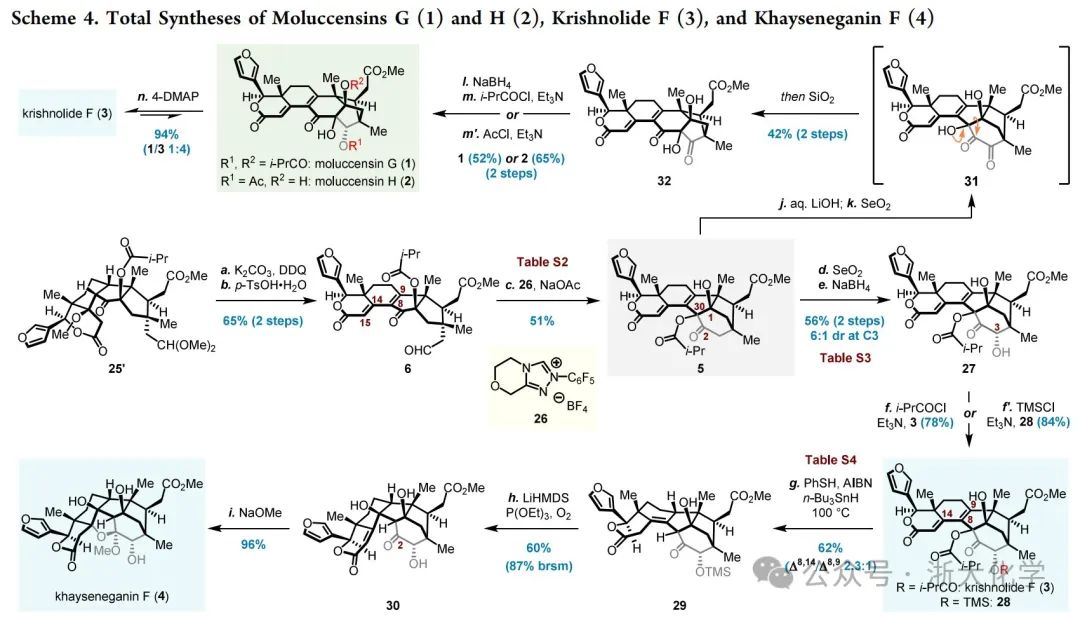
在获得足量的中间体5之后,作者开始着手moluccensins G (1)和H (2), krishnolide F (3)以及khayseneganin F (4)的合成。为立体选择性地引入α构型的C3位的羟基,作者首先尝试了直接的羰基α位羟基化反应,遗憾的是仅能得到β构型的C3羟基化的产物。为克服这一立体选择性问题,作者于此选择迂回的策略,先用SeO2对5进行Riley氧化,得到α-二酮(未展示)后再用NaBH4进行选择性还原,最终通过两步转化以56%的收率获得中间体27。随后,作者对新形成的C3位羟基进行酰化即可得天然产物krishnolide F(3)。后续khayseneganin F(4)的合成关键在于C30位的选择性去氧和α,β,γ,δ-不饱和酯的1,6-还原。然而,由27制备得到的TMS硅醚28在SmI2,L-Selectride,NiCl2/NaBH4,Stryker’s reagent,Cu(OAc)2/dppbz/PMHS,Mn(dpm)3/Ph(i-PrO)SiH2等一系列条件下均未能实现共轭还原。受López工作的启发,作者认为可以通过硫醇的共轭加成/自由基还原脱硫反应实现形式上的共轭还原。幸运的是,以甲苯为溶剂,底物28在80 ℃下与PhSH、AIBN、n-Bu3SnH反应,成功地实现了共轭还原和C30位的氧化态的脱除,得到了少量的发生1,6-还原反应的目标产物29(以1,4-还原反应为主)。升高温度至100 ℃时,1,6-还原反应的比例可得到有效的提高,最终以2.3:1的比例得到Δ8,14和 Δ8,9-单烯混合物,总产率为62%。随后,29在LiHMDS、P(OEt)3和O2的条件下立体选择性地引入C8位羟基。所得产物30在NaOMe促进下发生C2缩酮化/氧杂Michael加成串联反应,高效地实现了向天然产物khayseneganin F(4)的转化。
另一方面,中间体5经异丁酰基的水解和Riley氧化可得不稳定的α-二酮31。其可在硅胶柱层析过程中自发完成期望的C1(30→2)偶姻重排反应,最终以42%的总收率得到具有phragmalin型柠檬苦素骨架的32。最后,32经过立体选择性还原和酰化两步反应,分别以52%和65%的收率合成了moluccensins G(1)和H(2)。此外,作者进一步研究了moluccensin G(1)与krishnolide F(3)之间的仿生转化。室温下,moluccensin G(1)在4-DMAP的作用下会发生C1(2→30)偶姻重排,得到一对比例为1:4的moluccensin G(1)与krishnolide F(3)的混合物;然而在相同条件下,未能观察到krishnolide F(3)向moluccensin G(1)的重排反应。
总结
丁寒锋/宣军课题组从手性烯酮10和6-甲基环己-2-烯-1-酮(17)出发,经17-20步转化发散性地完成了两个phragmalin型柠檬苦素(moluccensins G和H)以及两个khayanolide型柠檬苦素(krishnolide F和khayseneganin F)的首次不对称全合成。关键步骤包括Liebeskind−Srogl偶联反应、中断型Nazarov环化反应、安息香缩合反应和双向的偶姻重排反应。该工作不仅简洁高效地合成了phragmalin和khayanolide型柠檬苦素,为两类柠檬苦素的模块化合成奠定了基础,还为这两种骨架之间的柠檬苦素间的生源转化提供了新的见解。
浙江大学化学系/化学前瞻技术研究中心饶培榕博士后、研究助理唐东民和化学系夏启东同学为本论文做出了主要贡献,丁寒锋教授和宣军研究员为共同通讯作者。本研究得到了化学系和国家自然科学基金的资助。
论文链接:https://pubs.acs.org/doi/epdf/10.1021/jacs.4c16265