
通过瞬态碳正离子复合物实现的不对称Hofmann-Löffler-Freytag型反应
背景介绍
自1899年被发现以来,碳正离子(carbenium ions)因其独特的电子结构和高反应活性一直是有机化学研究的核心课题之一。然而,如何实现对碳正离子参与的化学转化过程中化学选择性,区域选择性和立体选择性的精准调控,一直是该领域颇具挑战性的科学难题。虽然通过共振稳定或杂原子稳定的碳正离子通常可以实现较好的反应选择性,但缺乏此类稳定作用的碳正离子体系,其不对称转化反应仍鲜有报道。近年来,List(图1A)、Jacobsen(图1B)和Nelson等课题组通过调控烯烃质子化和C-X(X = Cl, O)键裂解产生的碳正离子中间体,成功实现了高对映选择性转化。在这些开创性工作中,手性催化剂通过与抗衡阴离子或试剂产生非共价相互作用来创造手性环境,从而实现了亲核试剂对碳正离子的对映选择性进攻。尽管取得了这些突破性进展为碳正离子化学开辟了新方向,但利用简单C(sp3)-H键直接作为碳正离子前体,进行不对称转化的研究仍处于起步阶段,其反应机制和选择性控制策略亟待深入探索。
惰性C(sp3)-H键由于其较高的键解离能(BDE)和较弱的极性特征,导致其活化过程面临巨大挑战。相较于传统的烯烃质子化和C-X键裂解过程,实现其选择性活化并高效转化为碳正离子更为困难。这一科学难题可追溯至1883年Hofmann发现的Hofmann-Löffler-Freytag(HLF)反应,该反应通过1, 5-氢原子转移(HAT)过程实现C(sp3)-H键的选择性活化。近十年来,研究者通过结合HAT和氧化自由基-极性交叉(ORPC)过程来产生碳离子中间体,并实现了C(sp3)-H键的官能团化反应。然而,该领域的发展仍面临显著挑战,特别是在其对映体选择性控制方面(图1C)。主要挑战包括:(1)前手性碳正离子中间体因其平面对称性导致其两个反应面难以有效区分,这降低了反应实现高对映体选择性的可能性;(2)碳正离子的高反应活性使得在无催化剂条件下,亲核进攻也能自发进行,从而产生难以抑制的外消旋背景反应;(3)碳正离子和手性催化剂之间的作用关系难以捉摸,特别是在氧化还原环境中。
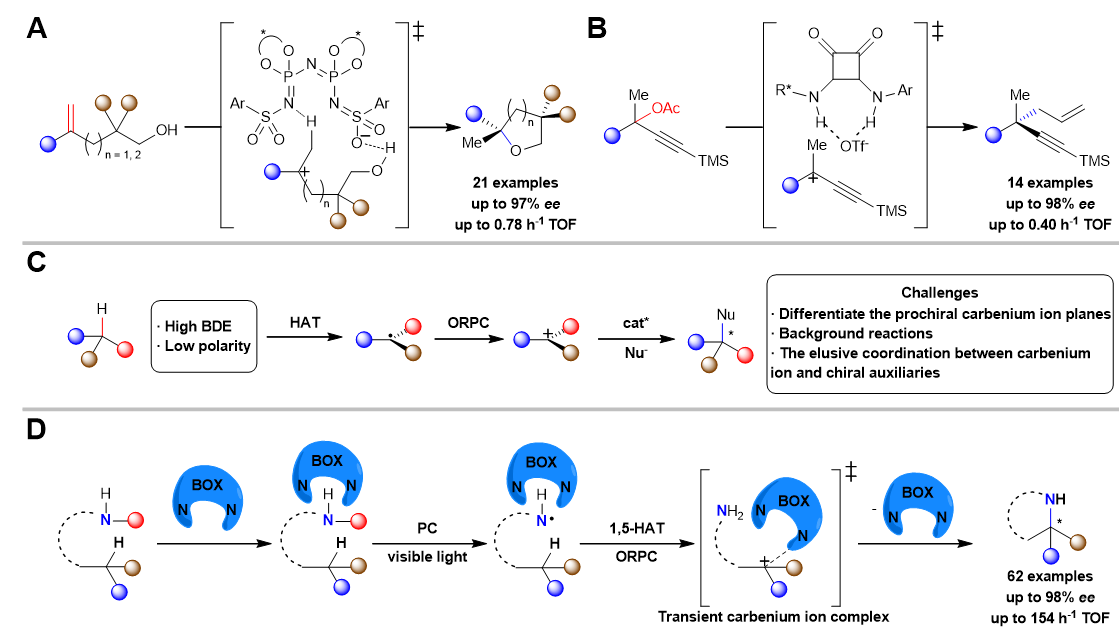
图1 碳离子对映体控制转化的研究进展
文章亮点
针对上述挑战难题,浙江大学陆展团队开发了一种新型的光/有机小分子协同催化不对称HLF型反应,使用手性双噁唑啉(BOX)作为有机催化剂,反应生成了具有优异对映选择性的手性Evans辅基(图1D)。该方案显示出高反应活性和对官能团的广泛容忍性。此外,与洪鑫团队合作进行了深入的机理研究和DFT计算,揭示了双噁唑啉催化剂一方面通过氢键作用活化底物来提升反应速率;另一方面通过与碳正离子中间体的瞬态配位作用来控制反应的对映选择性。
图文解读
在确定了反应最佳条件后,该课题组研究了C(sp3)-H键酰胺化反应的底物范围和局限性(图2)。实验证明,该反应条件与广泛的芳基取代基兼容,包括苯环邻位、间位和对位上的给电子和吸电子基团,以及其他官能团,如受保护的苯酚和茴香基。该反应可扩展到4 mmol,而产率和对映体选择性没有下降。此外,吲哚、噻吩、苯并呋喃等杂环化合物产生了相应的对映选择性良好的产物。最后,尿素衍生物也被证明是一个合适的底物。
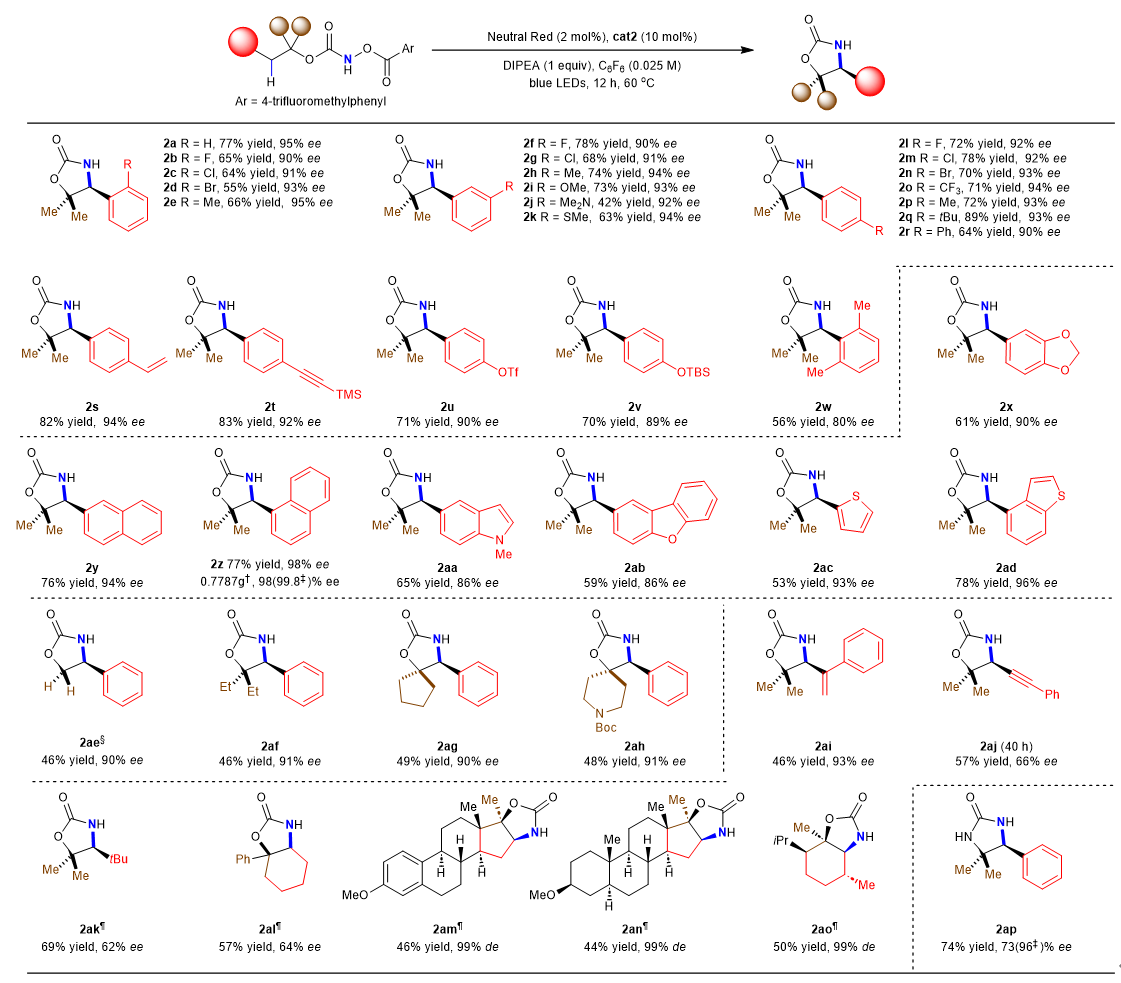
图 2 仲碳(sp3)-氢键的不对称酰胺化反应
通过使用BOX手性臂具有更富电子萘基的催化剂cat4,该方法还可以实现外消旋三级C(sp3)-H键的对映汇聚式酰胺化反应,从而构建季碳手性中心(图3)。当使用不同的给电子和吸电子取代苯基底物,反应也显示出较高的对映选择性。此外,该方法还可以用于构建手性螺环季碳中心。
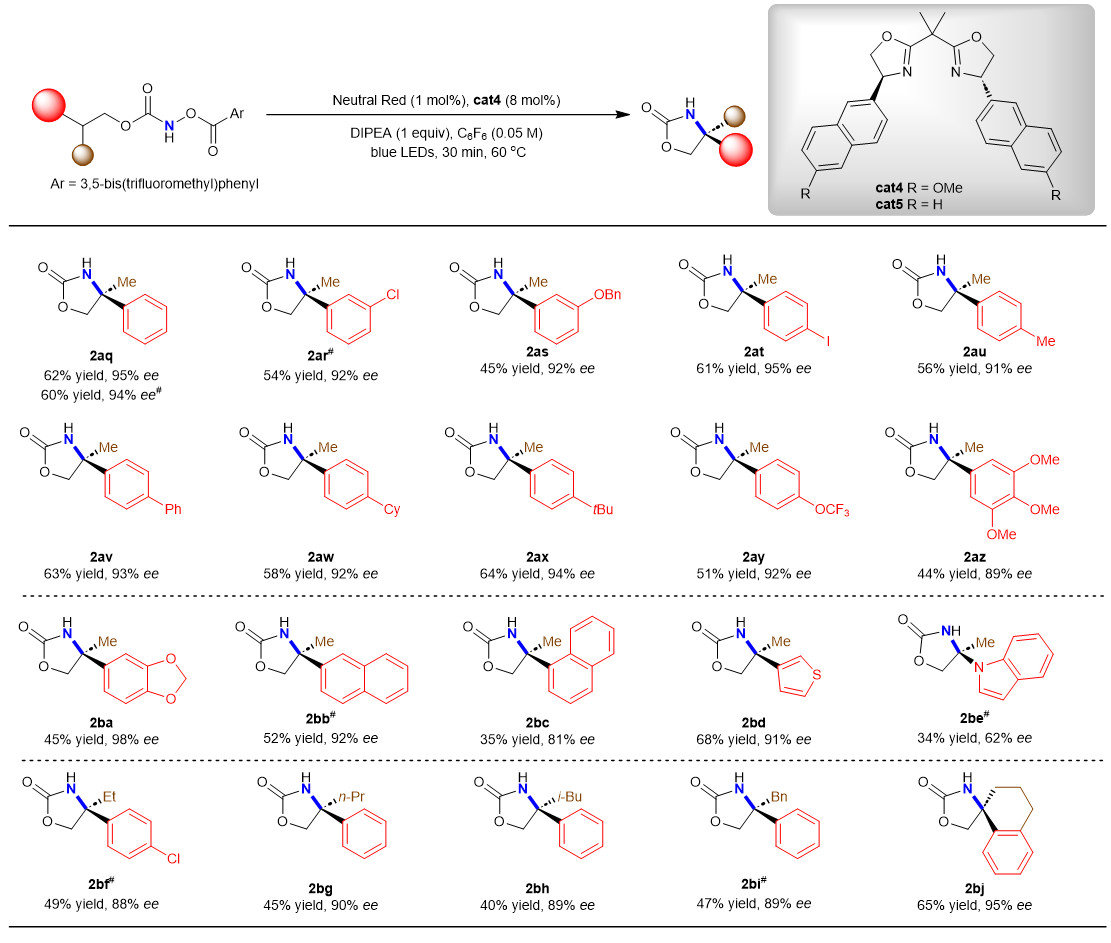
图3 叔碳(sp3)-氢键的对映体汇聚式酰胺化反应
作者将对照实验、同位素实验与DFT理论计算相结合,阐明该反应可能经历的催化循环如图4所示。首先光催化剂(PC)在蓝光照射下被激发为激发态PC*。底物1和cat2可以通过氢键相互作用形成复合物A,该复合物可被PC*物质还原,发生N-O键断裂形成氮自由基B。氮自由基B发生分子内1,5- HAT,产生苄基自由基C。该自由基又很容易被PC+氧化成苄基碳正离子D1。瞬态碳正离子复合物E可能是在BOX对苄位碳正离子中间体的热动力学捕获后生成的。通过DFT计算可以排除碳自由基被BOX捕获的途径。最终,该反应通过E的分子内对映选择性C-N键形成得到2a,并释放BOX。初步的理论研究详见原文。
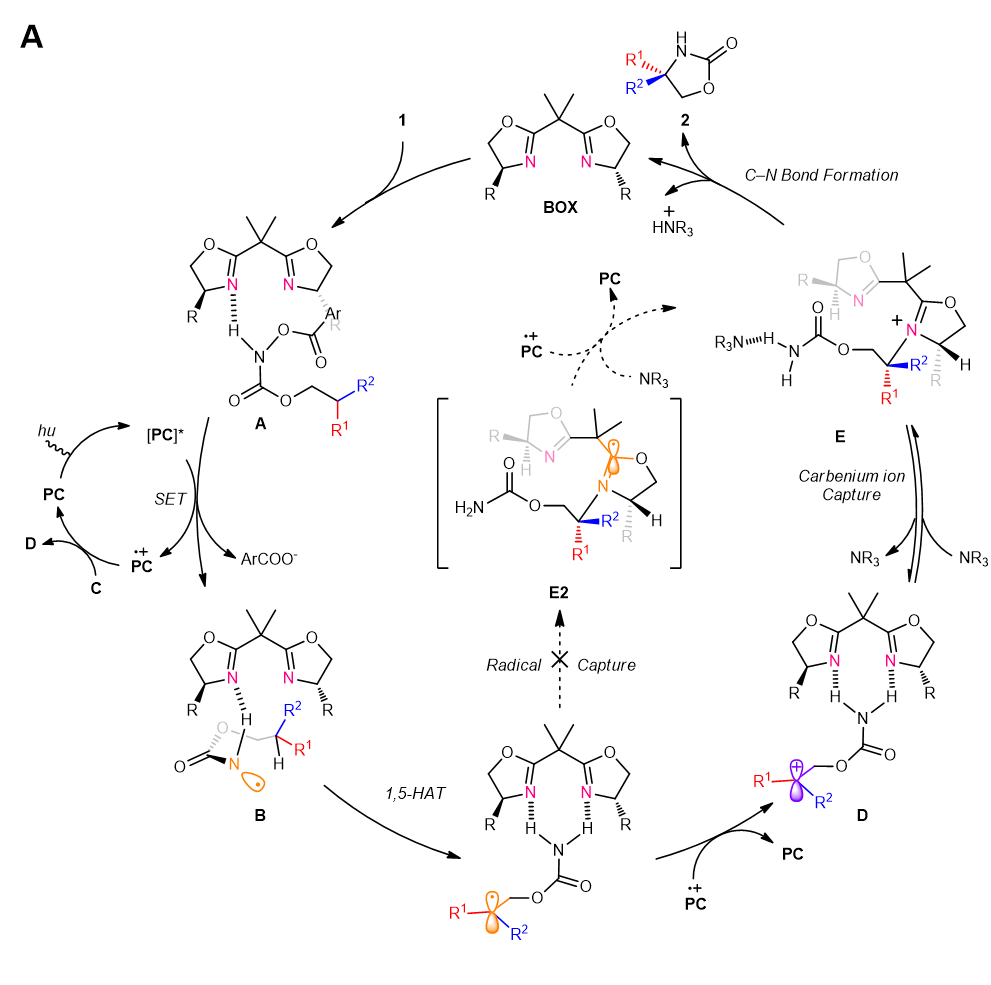
图4 可能机理
总之,陆展团队与洪鑫团队合作实现了可见光/手性双噁唑啉 (Box)协同催化的二级和三级碳氢键不对称酰胺化反应,构建具有重要合成价值的手性Evans辅基类产物,同时也提出了一个有效控制碳正离子不对称转化的催化模型。
文章链接:https://doi.org/10.1038/s41929-025-01329-2
课题组链接:https://person.zju.edu.cn/lu